This blog post covers some of the rationales that I put into when designing SkyMap, the project which involves making >400,000 sequencing runs accessible to everyone. This post could be informative to you when you are designing your next Big Data application in Bioinformatics. I have listed some of the problems that I faced and my rationales in solving them. In particular, this post will be focused on optimization to attain fast data retrieval with simple coding.
Fast data I/O from the Big -omic matrices and metadata
I chose the most debated Python Pandas pickle and NumPy format for the storing the data at the end as opposed to HDF5, Feather and relational database. The short story is because Python pandas pickle offers the fastest data reading speed as compared to HDF5 and Feather. The long story goes like this: Database performance optimizations revolve around trading off between data consistency, scalability, portability/accessibility, caching and read-write I/O performance.
- Caching in RAM: The reprocessed data volume is bigger than what can be easily fit and be cached into memory (RAM) comfortably.
- Problems:
- Fitting the data into the memory is key towards reducing both the code complexity and increase computation efficiency. The allelic read counts data is the one that I had the most trouble with as compared with expression data. The allelic read counts data has a high dimension as it is counting over genomic coordinate as opposed to counting at gene resolution.
- Comparing with HDF5 and Feather: The HDF5 in queryable format had a very poor compression rate and I/O rate.
- Solutions:
- I merged the allelic read count data into Python Pandas pickle chunks based on SRR IDs.
- The core idea behind efficient storage is about chunking the storages, which spans from CPU caching to distributed file storage optimization, and the beauty is that SRA IDs offer data locality as the NCBI SRA keeps the sequencing runs from the same study to have proximal IDs, and this data locality concord with the user data access behavior where they often query the data by study.
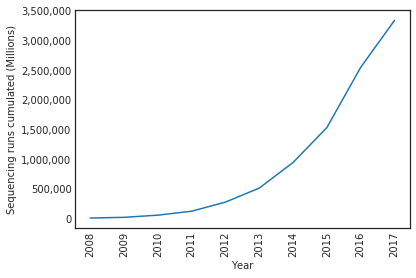
- Also, the portability issue has been greatly mitigated where we offer both the public JupyterHub and our conda environment. Also, since we store the data in AWS EFS which has high throughput and I/O. The cost of a complete data concatenation for each species and matrix takes only a couple of hours. In the end, the need for sophisticated scalability solution isn’t there, where the growth of sequecing data isn’t quite exponential yet (shown in the following plot), even assuming somehow the SRA data start doubling every two years within the next five years, concatenating the data from scratch will still complete within a day.
- We store the data using the memory mapped NumPy matrices. Memory mapping offers very efficient data access in the dense matrix, which can be wrapped around by Pandas Dataframe and works well with expression data.
- I merged the allelic read count data into Python Pandas pickle chunks based on SRR IDs.
- Problems:
- Data consistency:
- Solution: I use the NCBI IDs throughout without using my own so that if you have any question about the data you can look up the NCBI SRA website. In another, I simply try to avoid this problem by not creating the custom IDs like some major consortiums.
- Data and code maintainability
- Problem: The problem in academia is that we rarely have the financial resource to hire a software engineer to maintain the software.
- Solution: I hosted the JupyterHub on AWS with Kuberente and EFS which includes autoscaling, fault tolerance and complete resource monitoring, which save me the trouble of writing code nor hiring someone to babysit the JupyterHub. Each pipeline has been reduced down to a single notebook where a person just needs to hit “Run All”.
- Data portability and accessibility
- Problem: I and my colleagues all found existing custom databases difficult to use. For example, multiple huge consortiums only store their precious data in their customer database which you can query using only their custom REST/ Unix commands, which they could have totally just put the expression and metadata can fit into a simple 1MB CSV sheet. It means that each user needs to learn their lingos in order to retrieve the data.
- Solutions:
- We offer the JupyterHub to leverage the fact where Python offer a good data science ecosystem. JupyterHub allows the user to run their notebooks however they want in their own containerized environment. Also, the JupyterHub offer full transparency in terms of data retrieval, which is key in this bioinformatics world where it is filled with uncertainties.
- We packed all the metadata in a single pickle. It turns out that the SRA metadata, which covers the majority of the publicly available sequencing data on this planet, can fit into the memory of a single machine easily (<10GB) and be loaded in seconds.
- Keep the code as simple as possible. To maximize the code maintainability, I kept the codes as minimal as possible. In between documenting and reading thousand page documents versus succinct codes, I strongly prefer the later one, probably because of my past in complexity theory and teaching undergrads in CS classes, where the number of lines is often inversely correlated with the number of bugs. Also, the silo between computer language and computer language has been closing in recent years, many of my pure bio colleagues can understand and extend the Jupyter notebook with minimal comments, where they were able to extend the code without first learning all about software engineering. Also, all my Jupyter notebooks in the pipeline are linked together with the README.ipynb, where anyone in my lab can go and click through them to update the pipelines with thinking.
- Conda environment. Save me the trouble from keeping track of the package versioning.
- Why AWS EFS as opposed to S3 or Sage Synapse: The data was originally hosted on Sage Synapse in large because it is free, but a lot of the users I interviewed are having trouble with the custom commands from Synapse. S3 requires writing codes to manage it. The EFS storage system is much more expensive than S3 but it also has much fewer troubles. For example, I only need a simple UNIX rsync command to synchronize the data with EFS.
Just a disclaimer, most of my methods were un-orthodoxical and might only apply to my situation. Feel free to leave comments if you have any thoughts.
